Using this highly specialized scientific software you can analyze alignments of custom nucleotide or amino acid sequences in a phylogenetic framework.
PhyML
ver. 3.1
0,7 Mb (downloads: 1)
Update date:
11.08.2023
Developer:
Stephane Guindon and others
Windows version:
Windows XP
PhyML is a Windows tool oriented toward medical students and scientists. It allows users to study evolutionary history and relationships between separate organisms. Researchers can infer phylogenies under the maximum likelihood criterion to select the most probable tree given the data and a model of sequence evolution.
Brief description
The program is capable of handling various types of medical data such as DNA, RNA, protein, codon and binary sequences. Several different evolution models are supported. You can utilize the provided tree search algorithms and optimization methods to improve the output.
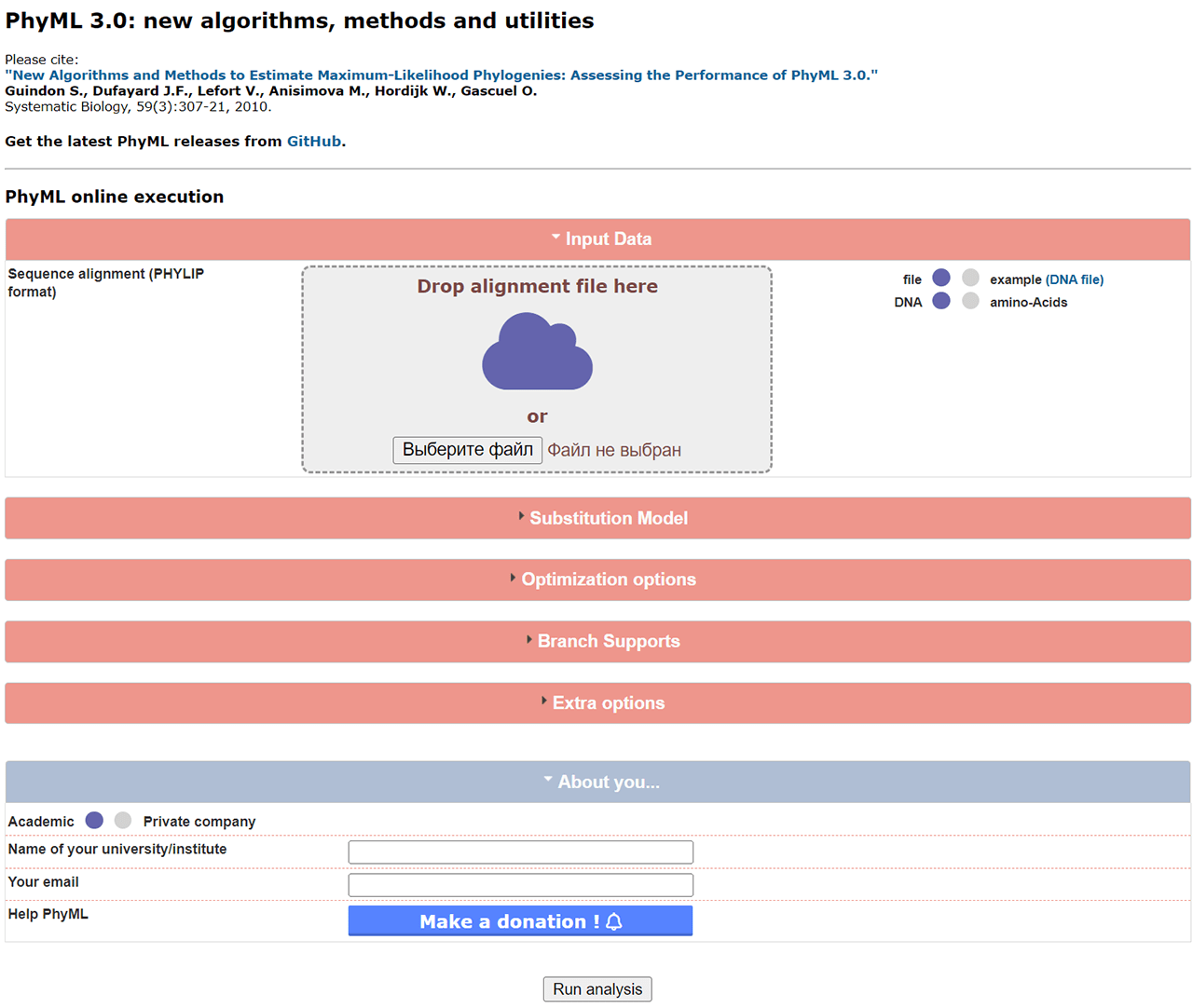
It is worth noting that PhyML is a command line utility. The full list of all possible arguments is located in the instruction manual. However, there is an online version of the service available on the official website. Users can upload source information and configure various processing parameters via a graphical web interface.
Other applications with similar functions like PAUP are available for download.
Statistical analysis
After importing a sequence alignment file in PHYLIP format you can choose the type of data and the number of sets to process. It is necessary to select the evolution model that fits the project better.
Users are able to compare several tree models by running likelihood ratio or AIC tests. Tools for exporting resulting files to the Newick or Nexus formats are provided as well.
Features
- free to download and use;
- helps medical researchers and students study relationships between organisms;
- supports various types of medical data such as DNA, RNA and binary sequences;
- you can choose the evolution model that better fit the project;
- compatible with all modern versions of Windows.
0,7 Mb (downloads: 1)