With the help of this highly specialized application scientific researchers can predict how small molecules bind to larger biomolecular targets such as proteins.
AutoDock
ver. 4.2.6
0,6 Mb (downloads: 18)
Update date:
30.12.2023
Developer:
The Scripps Research Institute
Windows version:
Windows XP
AutoDock is a Windows utility for molecular docking analysis. It can assist scientists in discovering new drug candidates by simulating their interactions with target proteins. Moveover, it is possible to scan massive libraries of molecules to identify those with promising properties.
Brief description
Using this tool you are able to assess the binding strength of different molecules by applying a semi empirical free energy force field. The attractive and repulsive forces between charged atoms are adjustable. Additionally, there is an option to evaluate hydrogen bonding, which is crucial for forming stable structures.
It is possible to predict hydrophobic interactions, where non polar molecules attract to each other to avoid water. Weak Van der Waals forces that occur due to electron fluctuations can be calculated as well.
Other solutions for molecular analysis such as MassLynx are available for download.
Docking process
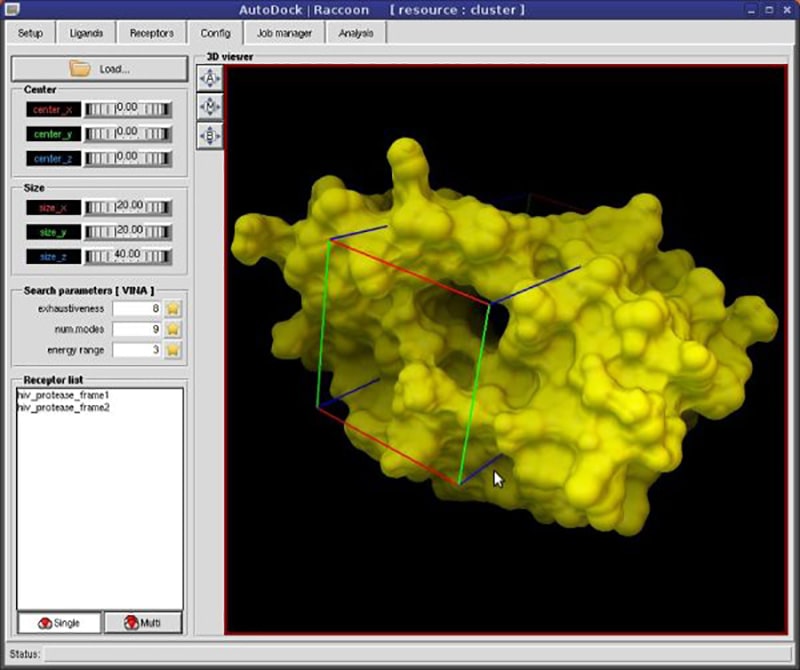
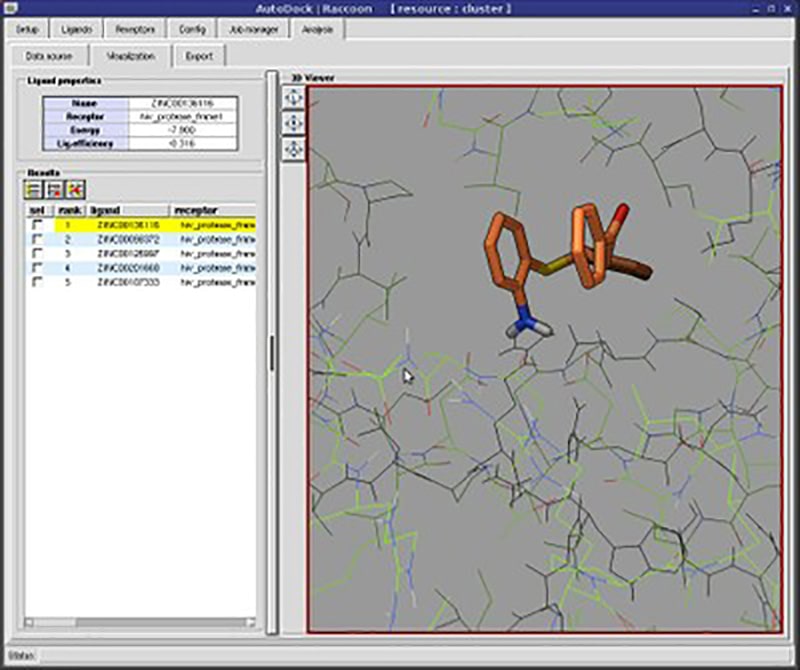
Users can view protein and ligand structures in 3D to visualize complex molecular structures. There are instruments for generating custom grids to define the search space within the receptor.
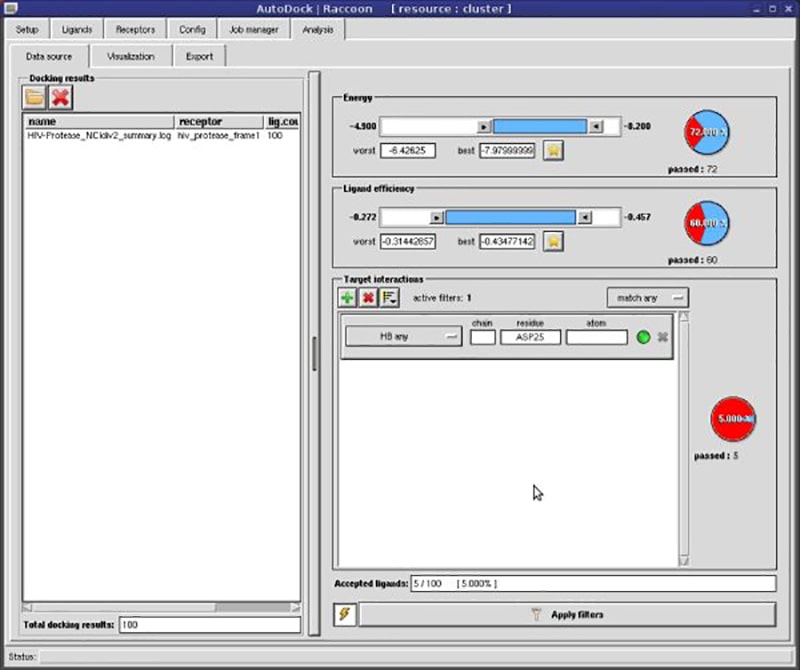
The application lets you generate detailed analysis reports. Resulting documents include all binding poses, energies and interactions observed during docking.
Features
- free to download and use;
- lets scientists assess the binding strength of various molecular structures;
- helpful for discovering new drugs and chemical compounds;
- it is possible to visualize protein and ligand connections;
- compatible with all modern versions of Windows.
0,6 Mb (downloads: 18)